Introduction
The National Institute on Drug Abuse (NIDA), to assist grantees conducting or planning to conduct clinical trials, has developed these guidelines for data and safety monitoring (DSM) plans, in accordance with NIH requirements. The purpose of the DSM plan is to ensure the safety of participants in clinical trials and the validity of trial results.
Grant applicants must submit a general description of the initial DSM plan as part of the research grant application. The Scientific Review Group will review the DSM plan and any comments or concerns will be included in the summary statement. A detailed DSM plan must be submitted to and approved by NIDA before the trial begins. The responsibility for compliance with the DSM plan rests with the grant recipient.
These guidelines do not take the place of Institutional Review Board (IRB) guidelines, Food and Drug Administration (FDA) requirements, or special NIH guidelines, e.g., NIH Guidelines for Research Involving Recombinant DNA Molecules. Specifically, Phase I and II gene transfer trials must comply with additional requirements imposed by NIH Guidelines, e.g., reporting of adverse events to the Office of Science Policy.
Background
In June of 1998, NIH issued a policy stating that “each Institute or Center (IC) in NIH should have a system for the appropriate oversight and monitoring of the conduct of clinical trials to ensure the safety of participants and the validity and integrity of the data for all NIH-supported or conducted clinical trials.” According to this policy, data and safety monitoring is required for all types of clinical trials, including physiologic, toxicity, and dose-finding studies (Phase I); efficacy studies (Phase II); efficacy, effectiveness and comparative trials (Phase III); etc. It includes all types of intervention studies, whether medication or non-medication (e.g., behavioral, prevention, diagnostic) trials. Monitoring should be commensurate with the study risks. This policy provides each IC with the flexibility to implement the requirement for data and safety monitoring as appropriate for its clinical research activities. (https://grants.nih.gov/grants/guide/notice-files/not98-084.html)
Further guidance to this policy (https://grants.nih.gov/grants/guide/notice-files/NOT-OD-00-038.html), released in June of 2000, stated that beginning with the October 2000 receipt date, investigators must submit a monitoring plan for Phase I and Phase II clinical trials to the IC before the trial begins. Also, a general description of data and safety monitoring plans must be included as part of the competing grant application.
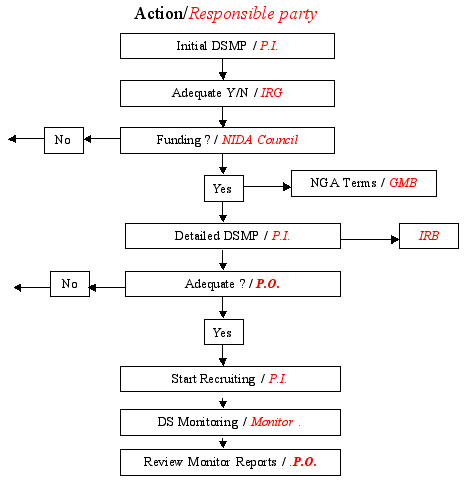
Trials that require a DSM Plan/Board
Figure 1 shows a decision tree to determine the need of a DSM plan and/or a DSM Board. According to the NIH DSM policies, DSM plans are required for all clinical trials of all phases for which grant support is sought. For the purpose of these guidelines, a clinical trial is a prospective study to test the effect of a biomedical or behavioral intervention in human subjects. It includes medications, herbal/nutritional supplements, physical interventions, behavioral interventions, prevention trials, and/or diagnostic tools that affect the outcome of the study participants.
The requirement of a DSM plan covers career and training awards in which the trainee has direct responsibility for conducting a clinical trial or in which award funds directly support a clinical trial.
Grants that include multiple clinical trials must submit a DSM plan to NIDA prior to initiation of each new trial. Initial funding of a grant and continuation of funding will be contingent on the Institute’s acceptance of the DSM plans.
In addition to the DSM plan, a Data and Safety Monitoring Board (DSMB) is generally required for Phase III clinical trials. For earlier medication trials (Phase I or II) and some behavioral clinical trials, a DSMB may be appropriate if the study has multiple clinical sites, is blinded, is First-in-Man, tests a high-risk intervention, or is conducted in vulnerable populations. Generally, Phase III trials involve larger numbers of subjects, but size is not a specific criterion for this designation.
The purpose of the DSMB is to monitor the safety of the interventions and the validity and integrity of the data from clinical trials that require a DSMB. The decision to establish a DSMB is commensurate with the level of risk and/or the number of treatment sites participating in the study. A DSMB may be required by the grantee’s institution, but the ultimate decision rests with the sponsoring Institute. In conducting its reviews and making recommendations, the DSMB works to assure that the safety of study subjects is protected while the scientific goals of the study are being met. In monitoring the data and safety of the trial, The DSMB may recommend continuation of the trial, modifications to the trial, or termination of the trial in the event of overwhelmingly significant efficacy difference between groups or unacceptable adverse events.
Adverse events are defined as any untoward medical occurrence that may present itself during treatment or administration of an intervention, and which may or may not have a causal relationship with the treatment. Serious adverse events are defined as any medical occurrence that results in death; is life-threatening; requires inpatient hospitalization or prolongation of existing hospitalization; creates persistent or significant disability/incapacity, or a congenital anomaly/birth defects. Adverse events and serious adverse events may occur in medication and non-medication trials. In non-medication trials, for example, AEs may include suicidal ideation/attempts, drug or alcohol overdose/withdrawal, etc. Guidance about definitions and reporting of AEs and SAEs can be obtained at:
- https://grants.nih.gov/grants/guide/notice-files/not99-107.html
- https://www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/
CDER/ContactCDER/default.htm - https://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/
Guidances/default.htm - https://www.fda.gov/Safety/MedWatch/default.htm
Figure 1. DSM Plan/Board Decision Tree

Elements of a DSM Plan
The overall elements of the DSM plan may vary depending on the potential risks, complexity, and nature of the trial. They also depend on whether it is an initial or a detailed DSM plan.
Initial DSM plan
Research grant applications that involve clinical trials must include a general description of the DSM plan. The PHS 398 application instruction indicates where to place this description and what to include. The initial DSM plan should include a brief description of data management methods, collection and reporting of Adverse Events (AE) and Serious Adverse Events (SAE), medical safety measures, communication plan with IRB/FDA/NIDA, interim analysis plans, and content and frequency of DSM reports to NIDA. The entity responsible for monitoring should be specified. The Scientific Review Group reviews the initial DSM plan and may provide comments or concerns, which are included in the summary statement. Appendix B, C, and D have samples of initial DSM plans for pharmacotherapy, behavioral, and prevention trials, respectively.
Detailed DSM plan
A detailed DSM plan must be submitted to and approved by NIDA before starting a new trial. Appendix A has a checklist of the basic elements of a detailed DSM plan. The responsibility for compliance with the DSM plan rests with the grant recipient. Notice of Grant Awards (NGA) will include terms to put into effect NIH DSM policies.
The detailed DSM plan should include:
Summary of the protocol
- Brief description of the protocol (Study design) and table for schedule of events
- Primary and secondary objectives and outcome measures
- Inclusion/exclusion criteria
- Power calculation and sample size
Trial Management
- List of participating enrolling clinics or data collection centers
- Planned enrollment timetable (graph showing time vs. projected enrollment)
(Enrollment: participants who are randomized and received treatment/intervention in the trial) - Target population distribution
Data Management and Analysis
- Data acquisition and transmission
- Data entry methods
- Data security and plan for protecting confidentiality
- Data analysis plan
Quality Assurance
- Procedures in place to ensure the validity and integrity of the data
- Procedures to guarantee the accuracy and completeness of the data, during data collection, entry, transmission, and analysis.
Regulatory Issues
- Reporting of SAEs
- Medication trials: to the IRB, NIDA, and, as applicable to the FDA.
- Non-medication trials: to the IRB and NIDA.
- Reporting of IRB actions to NIDA
- Report of changes or amendments to the protocol (Changes made to protocol must be pre-approved by NIDA)
Trial Safety
- Potential risks and benefits for participants
- Risk mitigation plan (Management of SAEs or other study risks)
- Trial stopping rules
- Process of AE/SAE collection, assessing by PI and/or medical monitor and reporting
- AE/SAE follow up plan
Trial Efficacy
- Plans for Interim Analysis of efficacy data (if applicable)
DSM Plan Administration
- Responsibility for data and safety monitoring
- Persons responsible for monitoring the trial
- Disclosure of any conflict of interest
- Frequency of DSM
- How often are the data reviewed in the course of the trial?
- Content of DSM report (to be submitted to NIDA PO annually)
- Brief description of the trial and progress
- Enrollment update and baseline sociodemographic characteristics
- Retention and disposition of study participants (active, completed, and terminated/withdrawn)
- Regulatory Issues (amendment, deviations, IRB report, Q.A issues)
- AEs and SAEs listings
- Efficacy (if applicable)
If applicable, DSM Board Plan
- Members and affiliation
- Conflict of interest
- Frequency of meetings
- Protection of confidentiality
- Monitoring activities (initial and ongoing study review)
- Communication plan to IRB, NIDA, and FDA (if applicable)
Review of DSM Plans by NIDA Staff
The Project Officer assigned to the grant will review the DSM plan prior to starting the trial. The review focuses on the quality of the process that the investigator is planning to have in place to ensure the safety of the participants and obtain reliable results. The Project Officer’s review is based on the adequacy with which the plan covers the essential elements outlined above. According to NIH policy, the detailed monitoring plan must be included as part of the protocol and submitted to the local IRB and reviewed and approved by the funding Institute and Center (IC) before the trial begins. The responsibility for compliance with the DSM plan rests with the grant recipient. Appendix E shows the review process for DSM related actions.
For questions or comments please contact:
Ivan D. Montoya, M.D., M.P.H.
Deputy Director, Division of Therapeutics and Medical Consequences
6001 Executive Blvd.
Bethesda, MD 20892-9551
Phone: (301) 827-5936
Fax: (301) 443-2599
e-mail: imontoya@mail.nih.gov
Appendix E. – DSM Plan Review Process