Details
Meeting Summary
The National Institute on Drug Abuse (NIDA) hosted an informational webinar to provide information and respond to questions from prospective applicants who may be interested in submitting grant applications in response to the funding opportunity (NOFO) PAR-21-183 - Developing Digital Therapeutics for Substance Use Disorders (UG3/UH3 Clinical Trial optional) - Webinar Notice - https://grants.nih.gov/grants/guide/notice-files/NOT-DA-21-052.html
Presentations
- Developing Digital Therapeutics for Substance Use Disorders - Pre-Application Webinar
Participating Agencies
- FUNDING- National Institute on Drug Abuse (NIDA)
- ADVISORY- Food and Drug Administration (FDA)
Funding Opportunity Purpose
This funding opportunity seeks to support
- Research to develop and test DTx for stand-alone treatments or integrated with FDA-approved SUD treatments;
- Specific DTx indications: prevention of SUD initiation, medication adherence, treatment retention, treatment of withdrawal, abstinence, or reduction of relapse
Digital therapeutics are clinical-grade mobile, web, or other software-based platforms designed to deliver treatments for medical conditions or diseases
These therapeutic interventions do not include wellness apps or telehealth which provides remote access to a clinician
PAR-21-183 is a U-Mechanism Cooperative Agreement
- Cooperative agreements are similar to R-mechanism awards
- In addition to a Program Officer, a NIDA Science Officer is assigned
- Science Officers actively work with the grantee to support the research. For example, this may include providing input on study design, interacting with FDA, evaluation of results and meeting of milestones and go/no-go criteria
UG3/UH3 – What is a Phased Award?
- The UG3/UH3 Cooperative Agreement involves 2 phases
- The UG3 phase, for up to 2 years, is designed to support a project with specific milestones and go/no-go criteria to be met by the end of the period
- The UH3 phase is to provide funding for up to 3 additional years following successful completion of the UG3
- UG3 projects that meet their milestones and go/no-go criteria will be administratively considered by NIDA and prioritized for transition to the UH3 phase
- Investigators submitting to this funding opportunity must address both UG3 and UH3 phases
UG3 Transition: Milestones and Go/No-Go Criteria
- Transition from the UG3 to UH3 phase is based on
- Meeting milestones and go/no-go criteria
- Programmatic priority
- Available funds
- The milestones and go/no-go criteria must
- Include quantifiable measures
- Be designed on what is needed to reach the next stage of development
Example Activities in the UG3 Phase
- Development of a finalized version of the DTx and validation in the study population;
- Evidence that the intervention effects efficacy related endpoints
- For example: craving, dependence, days of abstinence, etc;
- Evidence the intervention effects behavioral endpoints as they relate to SUD
- For example: measures of working memory, impulsivity, risk-taking propensity, distress tolerance, self-regulation, stress reactivity, etc;
- Evidence an adequate dose range/treatment duration for the DTx intervention(s) can be applied with acceptable safety, tolerability, adherence, etc;
- Evidence when integrated with an FDA-approved treatment, there is enhanced adherence, retention, efficacy or effects on other measures relevant to the approved treatment;
- Completion of a proof-of-concept, feasibility clinical trial;
- Q-submission to obtain FDA feedback on the regulatory pathway;
- Completion of IDE-enabling studies;
- Filing an IDE;
- Approval of an IDE or confirmation that an IDE is not required
UH3 Phase
- Funding for the UH3 phase is contingent on successfully meeting the milestones in the UG3 phase.
- The UH3 phase supports the next step in the development of the intervention and progression towards FDA authorization.
- Applicants are expected to describe the entry and exit points of the proposed research in the development continuum.
- The application should provide quantifiable milestones to determine success of the UH3.
Working with the FDA
- Investigators are strongly encouraged to contact the FDA Center for Devices and Radiological Health (CDRH) via the Pre-Submission process to discuss the proposed development pathway and clinical validation data requirements
- See FDA presentation (below)
Administrative Review by NIH Staff
Submissions evaluated by NIH staff for:
- Completeness
- Presence of both UG3 and UH3 phase
- Presence of required application sections
- Biosketch requirement (NOT-OD-21-073)
- Clinical trial information (for clinical trial applications)
- Adherence to NIH application policies (Appendix, Human Subjects, etc.)
- Funding opportunity requirements
- Presence of milestone and timeline sections
- Adherence to the prescribed direct budget cost for the UG3 phase (500k/yr.)
- Duration of the UG3 phase (2 years. max)
Incomplete applications or applications not adhering to the funding opportunity requirements may be returned without review
Peer Review Panel
- Peer review of applications conducted at NIDA
- Special Emphasis Panel created by the SRO based on the areas of science described in the application
- Conflicts of interest are managed for all panel members
- At least three reviewers assigned to evaluate each application
- Panel will receive guidance from NIH staff on how to evaluate the application
- Panel members have at least 30 days to evaluate the application
- Meeting roster is publicly available 30 days before the meeting
Application Review Criteria – Section V of Funding Opportunity (NOFO)
Core Review Criteria
- Significance
- Investigators
- Innovation
- Approach
- Environment
Additional Review Criteria
- Study Timeline (for CT application)
- Protection of Human Subjects
- Inclusion of Women, Minorities, and Individuals Across the Lifespan
- Resubmission
Overall Impact
- Likelihood for the project to exert a sustained, powerful influence on the research field(s) involved
Additional Review Considerations
- Resource Sharing Plan
- Budget
- Foreign Organizations
Review Criterion Details
Significance
- Addresses an important problem or a critical barrier to progress in the field
- Rigor of prior research that serves as the key support for the proposed project
- Contribution to scientific knowledge, technical capability, and/or clinical practice
Investigators
- Experience, Expertise, Accomplishments, and Leadership plan
- Funding opportunity specific: Methodological and statistical expertise to evaluate digital intervention design, user engagement, and functional outcomes
Innovation
- Novelty of proposed project – concept, methodologies, intervention, invention, new application
Environment
- Institutional support, physical resources, collaborative arrangements
- Funding opportunity specific: Support for timely subject recruitment and completion of both phases
Approach
- Establishes feasibility, strategies for a robust and unbiased approach Addresses potential problems, alternative strategies, risk management, and benchmarks for success Considers biological variables and protection of human subjects
- Funding opportunity specific:
- Regulatory pathway & requirements for product approval
- Appropriateness and adequacy of milestones and go/no-go criteria
- Quality of the proposed product design and preliminary data
- Quality of measures proposed for user engagement/adherence, efficacy or behavioral endpoints
- Sufficiency of data to evaluate efficacy for reduced substance use or abstinence
Peer Review Outcome
- As part of the scientific peer review, all applications will receive a written critique (summary statement)
- Applications may undergo a selection process in which only those applications deemed to have the highest scientific and technical merit (generally the top half of applications under review) will be discussed and assigned an overall impact score
- Summary statement will be available within 4-6 weeks after the peer review meeting
Award and Eligibility Information
- Award Information:
- The funding opportunity will support New Applications
- The funding opportunity is Clinical Trial Optional – accepting applications that either propose or do not propose clinical trials (as specified in the funding opportunity)
- Application budgets are limited to $500,000 direct costs per year in UG3 phase
- Unlimited budget in UH3 phase but must reflect actual needs of the proposal
- The project period is limited to 2 years for the UG3 and 3 years for UH3 phases
- Eligibility:
- Refer to the funding opportunity for specific information on eligible applicants, eligible individuals, required registrations, and other eligibility information
Key Dates for 2021 Submissions
- Letter of Intent Due Date: July 2, 2021; November 6, 2021
- Application Due Date: August 4, 2021; December 6, 2021
- AIDS Application Due Date: Not applicable to this funding opportunity
- Scientific Merit Review: November 2021; March 2022
- Advisory Council Review: January 2022 ; May 2022
- Earliest Start Date: April 2022; July 2022
Key Contacts
- Program Officials
- Will M. Aklin, NIDA – aklinwm@mail.nih.gov
- Kevin Walton, NIDA – kevin.walton@nih.gov
- Review
- Dharmendar Rathore, NIDA - dharmendar.rathore@nih.gov
- Grants Management
- Pamela Fleming, NIDA – pfleming@mail.nih.gov
- Interacting with FDA and considerations for Digital Therapeutics - Developing Digital Therapeutics for Substance Use Disorders
Outline
- FDA guidance documents
- Early interaction with FDA
- Investigational device exemptions and study risk determinations
- Points to consider for digital therapeutic device intended for SUD
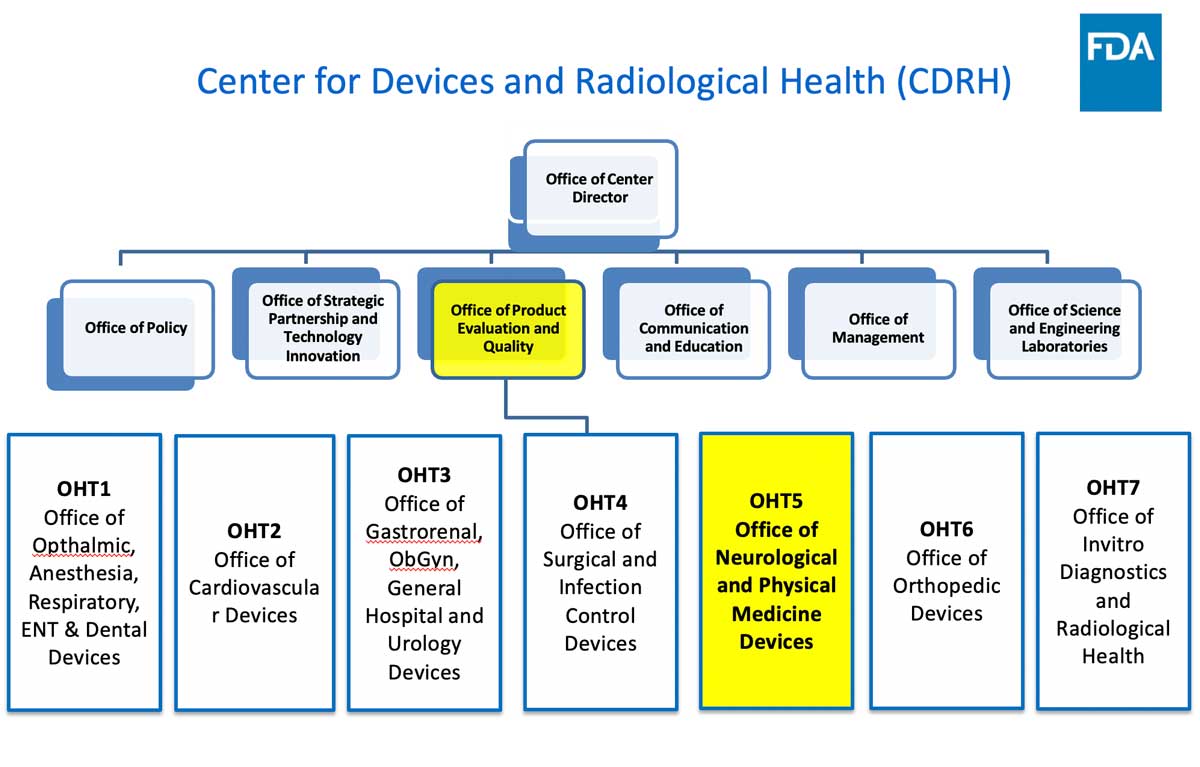
- FDA/CDRH/OHT5 overview and organization
FDA Guidance Documents
- General Wellness Guidance Document
- Policy for Device Software Functions and Mobile Medical Applications
- Significant Risk/Non-Significant Risk Guidance Document
- FDA and Industry Procedures for Section 513(g) Requests for Information under the Federal Food, Drug, and Cosmetic Act
- “Software as a Medical Device (SAMD): Clinical Evaluation”
- Benefit Risk Guidance for IDE Submissions
- Benefit-Risk Guidance for Medical Device Premarket Approval and De Novo Classifications
- Benefit-Risk Guidance for 510(k) Submissions
- IDE Submission Information
- Design Considerations for Pivotal Clinical Investigations Guidance
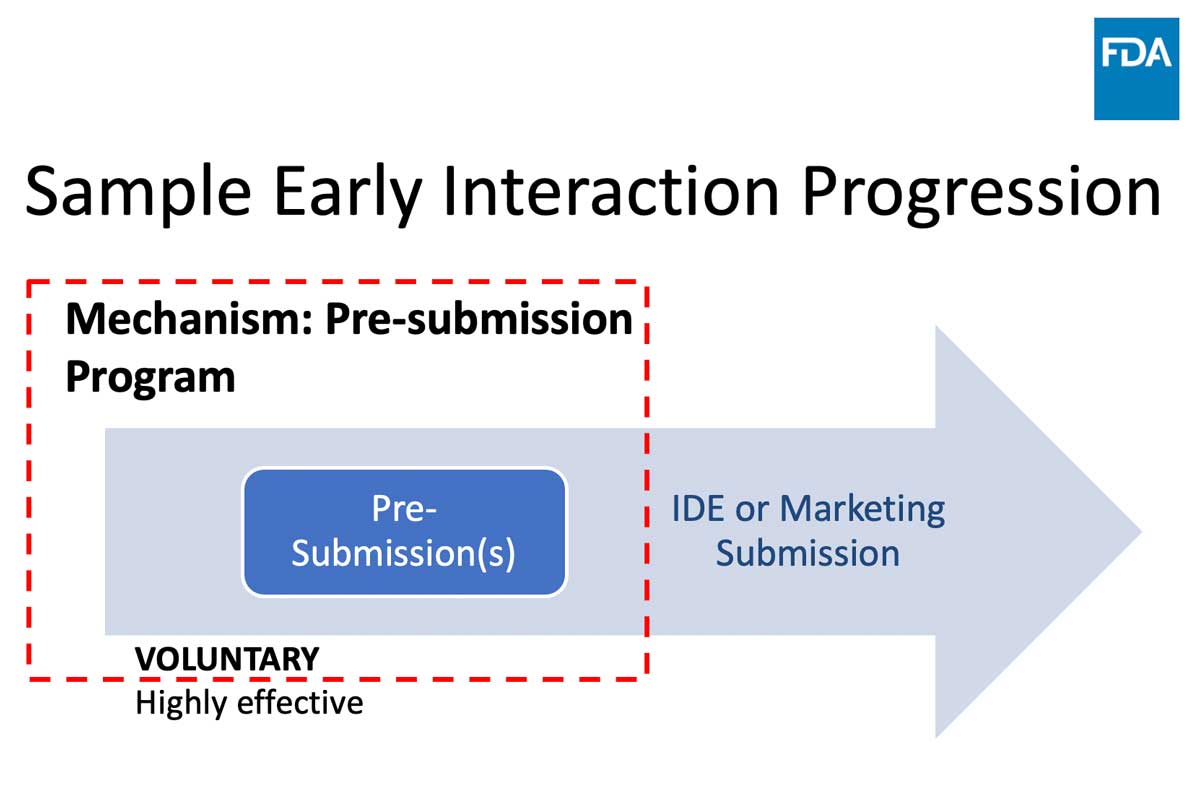
Image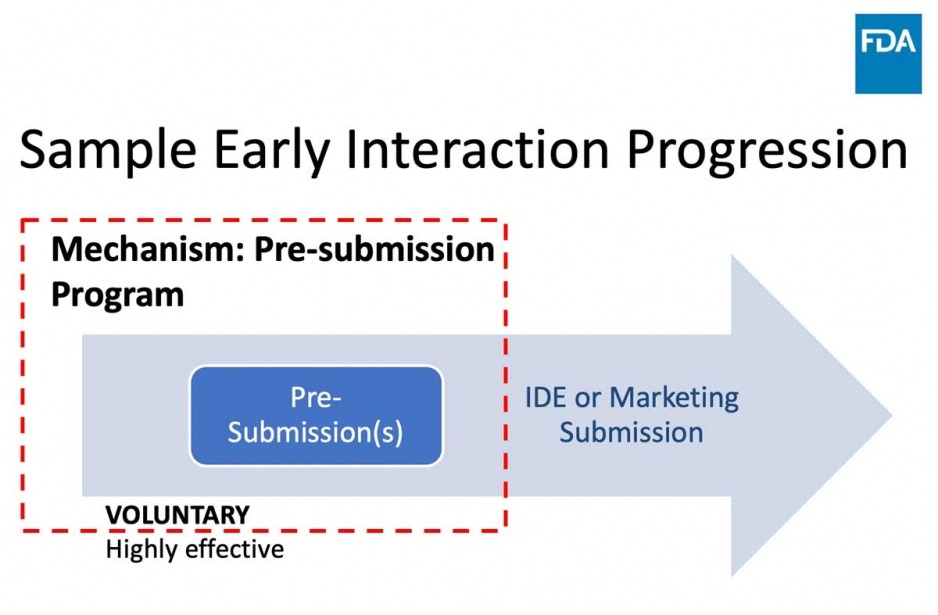
Q-Submissions
WHAT: an opportunity to obtain FDA feedback prior to IDE or marketing submission
WHAT YOU GET: Written feedback and an optional meeting
WHAT IT IS NOT: Pre-submissions are NOT intended for “pre-review” of data
The purpose of any discussion during the meeting is to clarify our feedback, not to respond in real-time to new information or proposals.Guidance Document
When is an IDE Needed?
- When a device is a significant risk device.
- 21 CFR 812.3(m) defines “Significant Risk” as and investigational device that:
- Is intended as an implant and presents a potential for serious risk to the health, safety, or welfare of a subject;
- Is purported or represented to be for a use in supporting or sustaining human life and presents a potential for serious risk to the health, safety, or welfare of a subject;
- Is for a use of substantial importance in diagnosing, curing, mitigating, or treating disease, or otherwise preventing impairment of human health and presents a potential for serious risk to the health, safety, or welfare of a subject; or
- Otherwise presents a potential for serious risk to the health, safety, or welfare of a subject.
Study Risk Determinations
WHAT IT IS: an opportunity to obtain FDA feedback on whether your proposed study is a significant risk or non-significant risk study
WHAT YOU GET: Written letter outlining FDA’s determination of your proposed study as significant risk or non-significant risk
Guidance Document
Image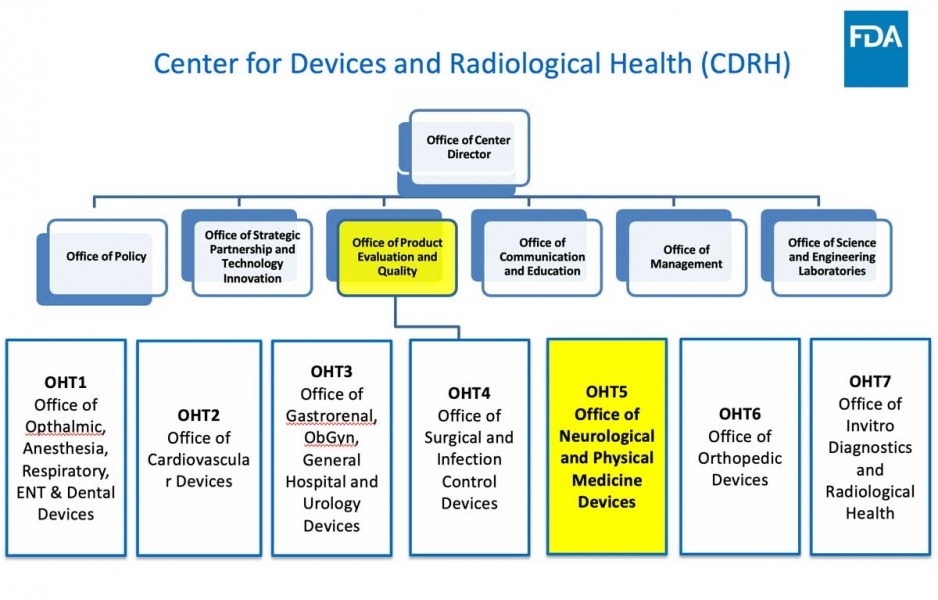
www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpcd/classification.cfm
Office of Health Technology 5 (OHT 5: Neurological and Physical Medicine Devices) Office Director Christopher Loftus, M.D. (Acting) 301-796-4377 Deputy Office Director John Marler, M.D. 301-796-4221 Deputy Office Director CAPT Nina Mezu-Nwaba 301-796-5494 Chief Medical Officer Christopher Loftus, M.D. 301-796-4377 Associate Director Vacant Associate Director for Operations Tonja Galberth 301-796-5959 Associate Director for Professional Development Vacant Safety Signal Manager Sonja Sokic 240-402-3679 Division of Health Technology 5 A (Neurosurgical, Neurointerventional and Neurodiagnostic) Division Director Xiaolin Zheng, Ph.D. 301-796-2823 Assistant Director for Neurosurgical Devices Adam Pierce 240-402-6128 Assistant Director for Neurointerventional Devices Naira Muradyan 240-402-4098 Assistant Director for Neurodiagnostic Devices Jay Gupta 301-796-2795 Division of Health Technology 5 B (Neuromodulation and Rehabilitation Devices) Division Director Vivek Pinto, Ph.D. 301-796-6610 Assistant Director for Neurostimulation-Neurology Devices Patrick Antkowiak, Ph.D. (Acting) Assistant Director for Neuromodulation-Psychiatry Devices Pamela Scott 301-796-5433 Assistant Director for Acute Injury Devices Heather Dean, Ph.D. 240-402-9874 Assistant Director for Neurodegenerative Devices Amber Ballard, Ph.D. 240-402-9983 Point of Contact for General Submission Questions
DICE - Division of Industry and Consumer Education
EMAIL : DICE@fda.hhs.gov
Phone: 1(800) 638-2041 or (301) 796-7100
Press 1 to speak to the Consumer Team
Press 2 to speak to the Industry Team